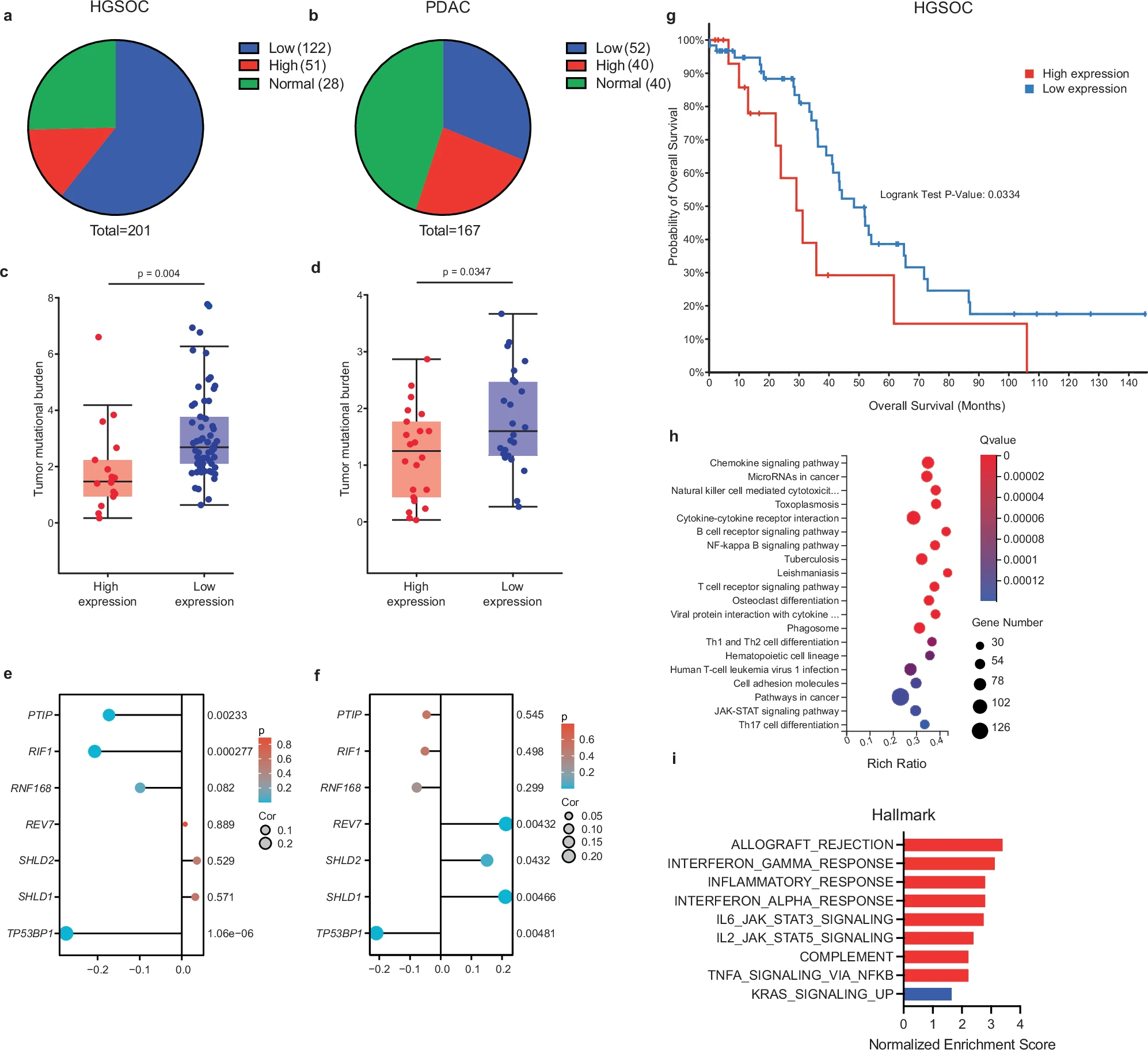
Zaburzenia molekularnych mechanizmów naprawy uszkodzeń DNA stanowią podstawę nowoczesnych metod leczenia nowotworów. Białko 53BP1 reguluje wybór między dwoma podstawowymi sposobami (tzw. ścieżkami) naprawy dwuniciowych uszkodzeń DNA: naprawą poprzez scalanie niehomologicznych końców DNA (ang. non-homologous end joining, NHEJ) oraz rekombinacją homologiczną (ang. Homologous recombination, HR). Ścieżka HR nie działa prawidłowo w komórkach z mutacjami genów BRCA1/2, co czyni je wrażliwymi na inhibitory białka PARP. Istotne wyzwanie terapeutyczne stanowi jednak oporność na inhibitory PARP, którą komórki z mutacjami BRCA1/2 mogą rozwijać m.in. poprzez supresje genów uczestniczących w regulacji naprawy DNA, głównie genu TP53BP1.
Najnowszy artykuł opublikowany przy współpracy naszej doktorantki Zuzanny Nowickiej oraz prof. Wojciecha Fendlera z zespołem prof. Dipanjana Chowdhury pokazuje, że utrata 53BP1 powoduje w komórkach aktywację ścieżki cGAS-STING i sygnałów prozapalnych, co z kolei prowadzi do wzrostu infiltracji guza przez cytotoksyczne limfocyty T i aktywację innych składowych odporności przeciwnowotworowej. Choć mechanizm, w którym czynnik uszkadzający materiał genetyczny lub zapobiegający jego prawidłowej naprawie aktywuje odpowiedź immunologiczną skierowaną przeciw nowotworowi nie jest nowy (w projekcie finansowanym z grantu PRELUDIUM BIS NCN Zuzanna Nowicka pod opieka prof. Wojciecha Fendlera bada na przykład, w jaki sposób chemioadioterapia może zmieniać środowisko immunologiczne raka płuca), po raz pierwszy wykazaliśmy że utrata 53BP1 uwrażliwia nowotwór na działanie immunoterapii. Może to w przyszłości stanowić podstawę do personalizacji terapii przeciwnowotworowej.
Zachęcamy do zapoznania się z pełnym tekstem artykułu, dostępnym w otwartym dostępie tutaj.
Nasza najnowsza publikacja w Nature Communications!